Mm9 multiple alignment: Difference between revisions
| Line 692: | Line 692: | ||
</TR> | </TR> | ||
</TABLE> | </TABLE> | ||
==Additional doBlastzChainNet.pl parameters== | |||
These parameters for blastz are placed in the DEF file used by the doBlastzChainNet.pl perl script. There are some additional parameters worth discussing: | |||
1. SEQ1* variables describe the target/reference assembly, while SEQ2* variables describe the query assembly. | |||
If those are the same assembly, then we're doing self-alignments and will drop aligned blocks that cross the diagonal. | |||
2. SEQn_CHUNK and SEQn_LAP determine the step size and overlap size of chunks into which large target or query sequences are to be split before alignment. | |||
According to reasons explained in the blastz paper, SEQ1_LAP is lways set to 10000, while SEQ2_LAP is traditionaly set to 0 | |||
3. SEQn_LIMIT decides what the maximum number of sequences should be for any partitioned file (the files created in the tParts and qParts directories). | |||
This limit only effects SEQ1 or SEQ2 when they are 2bit files. Some 2bit files have too many contigs which will result in blastz hippos (jobs taking | |||
forever compared to the other jobs). SEQ1_LIMIT defaults to 30 and SEQ2_LIMIT defaults to 100. By setting a lower limit, hippos may be avoided but | |||
more jobs will result. By setting this limit higher, fewer, longer running jobs result. | |||
The specifics of the target and query genomes may result in very different blastz run sizes, but CHUNK and LIMIT can be used to tune the size of the blastz run. | |||
One means of tuning is to run <CODE>doBlastzChainNet.pl</COCDE> with the <CODE>-stop=partition</CODE> flag to test the results of the partition step until satisfied, | |||
then continue with the <code>-continue=blastz</code>. To determine the number of blastz jobs that will result from a given partition, multiply the number of lines in | |||
<i>target</i>.lst by the lines in the <i>query</i>.lst. | |||
==default blastz parameters== | ==default blastz parameters== | ||
Revision as of 23:21, 11 April 2008
Mouse mm9 multiple alignment/conservation track
|
To avoid artifacts in downstream processing of the UCSC multiple alignments, it is important to be careful on the use of the parameters used in the blastz processing pipeline. There are a number of steps in the pipeline and a variety of tunable parameters involved. This page will track the various parameters used in the alignments as they proceed toward the completion of a multiple alignment conservation track on the mm9 mouse (NCBI build 37) assembly The chrom count in the table below does not include haplotypes, chr*_random, chrUn or chrM unless chrUn or scaffolds are the only sequences for that assembly. The genome size has the same limitation as the chrom count, no randoms. Tree distances are from the hg18 28-way measurements, with ponAbe2 and calJac1 manually inserted into the tree. 1 Mb = 1000 * 1000 = 1,000,000 bases Types of mafs used in the multiple alignment:
|
 30-way phylogenetic tree |
axtChain parameters and end results
xx.4| name db | chrom count (*) |
genome size |
tree distance |
axtChain minScore |
axtChain linearGap |
% of mm9 matched |
% of other matched by mm9 |
done |
|---|---|---|---|---|---|---|---|---|
| mouse mm9 self alignment |
21 | 2654 Mb | 0.0 | 10000 | medium | 14.458 | N/A | 2007-08-31 |
| rat rn4 | 21 | 2718 Mb | 0.160657 | 5000 | medium | 65.380 | 66.538 | 2007-08-31 |
| human hg18 | 24 | 3080 Mb | 0.452619 | 3000 | medium | 38.499 | 35.201 | 2007-08-16 |
| Rhesus rheMac2 | 22 | 2864 Mb | 0.452745 | 3000 | medium | 38.087 | 41.335 | 2007-09-07 |
| Orangutan ponAbe2 | 25 | 3029 Mb | 0.453809 | 3000 | medium | 34.902 | 30.659 | 2007-09-21 |
| Marmoset calJac1 | 49724 | 3029 Mb | 0.454272 | 3000 | medium | 32.971 | 30.302 | 2007-09-07 |
| Chimp panTro2 | 25 | 3175 Mb | 0.454514 | 3000 | medium | 37.674 | 34.269 | 2007-09-25 |
| GuineaPig cavPor2 | 295514 | 3403 Mb | 0.479871 | 3000 | medium | 18.326 | N/A | 2007-09-21 |
| Horse equCab1 | 32 | 2056 Mb | 0.479871 | 3000 | medium | 34.782 | 37.217 | 2007-09-25 |
| TreeShrew tupBel1 | 150851 | 3660 Mb | 0.494934 | 3000 | medium | 21.099 | N/A | 2007-10-01 |
| Bushbaby otoGar1 | 120882 | 3420 Mb | 0.498957 | 3000 | medium | 22.972 | N/A | 2007-09-28 |
| Armadillo dasNov1 | 304391 | 3856 Mb | 0.517360 | 3000 | medium | 16.547 | N/A | 2007-10-02 |
| Rabbit oryCun1 | 215471 | 3464 Mb | 0.519779 | 3000 | medium | 18.945 | N/A | 2007-09-29 |
| Cat felCat3 | 217790 | 4045 Mb | 0.530610 | 3000 | medium | 19.077 | N/A | 2007-09-29 |
| Dog canFam2 | 39 | 2445 Mb | 0.533544 | 3000 | medium | 32.362 | 34.891 | 2007-09-05 |
| Elephant loxAfr1 | 233134 | 3707 Mb | 0.536627 | 3000 | medium | 18.052 | N/A | 2007-10-01 |
| Cow bosTau3 | 30 | 2434 Mb | 0.540852 | 3000 | medium | 26.352 | 25.909 | 2007-09-25 |
| Hedgehog eriEur1 | 379801 | 3367 Mb | 0.632457 | 3000 | medium | 10.022 | N/A | 2007-10-02 |
| Shrew sorAra1 | 262057 | 2936 Mb | 0.658734 | 3000 | medium | 9.556 | N/A | 2007-10-01 |
| Tenrec echTel1 | 325491 | 3823 Mb | 0.666303 | 3000 | medium | 11.141 | 14.144 | 2007-09-27 |
| Opossum monDom4 | 9 | 3431 Mb | 0.909852 | 5000 | loose | 9.752 | 7.254 | 2007-09-27 |
| Platypus ornAna1 | 201522 | 1996 Mb | 1.165888 | 5000 | loose | 5.417 | 7.359 | 2007-09-25 |
| Chicken galGal3 | 33 | 1032 Mb | 1.285399 | 5000 | loose | 3.729 | 8.152 | 2007-09-25 |
| Lizard anoCar1 | 7233 | 1781 Mb | 1.404225 | 5000 | loose | 3.406 | 4.934 | 2007-09-21 |
| X. tropicalis xenTro2 | 19759 | 1513 Mb | 1.726205 | 5000 | loose | 3.131 | 2.651 | 2007-09-24 |
| Stickleback gasAcu1 | 21 | 400 Mb | 2.012649 | 5000 | loose | 1.849 | 9.791 | 2007-09-07 |
| Zebrafish danRer5 | 25 | 1547 Mb | 2.027153 | 5000 | loose | 3.255 | 4.625 | 2007-09-13 |
| Tetraodon tetNig1 | 21 | 217 Mb | 2.051015 | 5000 | loose | 1.763 | 12.341 | 2007-09-07 |
| Fugu fr2 | 1 | 400 Mb | 2.086669 | 5000 | loose | 1.794 | 10.784 | 2007-09-07 |
| Medaka oryLat1 | 24 | 724 Mb | 2.200402 | 5000 | loose | 1.933 | 6.495 | 2007-08-31 |
(*) chrom count does not include haplotypes, chr*_random, chrUn or chrM unless chrUn or scaffolds are the only sequences for that assembly.
The genome size has the same limitation as the chrom count, no randoms.
Tree distances are from the hg18 28-way measurements, with ponAbe1 and calJac1 manually inserted into the tree.
blastz alignment parameters details
One special run, Rat rn4:
M=40M, K=4500, L=4500, Y=15000, T=2 O=600, Q=mouse_rat.q E=5540
| query | abridged repeats |
M | K | L | Q | Y |
|---|---|---|---|---|---|---|
| Rat rn4 | yes | 40M | 4500 | 4500 | mouse-rat | 15000 |
| Human hg18 | yes | 40M | 3K | 3K | default | 9400 |
| Rhesus rheMac2 | yes | 40M | 3K | 3K | default | 9400 |
| Orangutan ponAbe2 | no | 50 | 3K | 3K | default | 9400 |
| Marmoset calJac1 | no | 50 | 3K | 3K | default | 9400 |
| Chimp panTro2 | yes | 40M | 3K | 3K | default | 9400 |
| GuineaPig cavPor2 | no | 50 | 3K | 3K | default | 9400 |
| Horse equCab1 | no | 40M | 3K | 3K | default | 9400 |
| TreeShrew tupBel1 | no | 50 | 3K | 3K | default | 9400 |
| Bushbaby otoGar1 | no | 50 | 3K | 3K | default | 9400 |
| Armadillo dasNov1 | no | 50 | 3K | 3K | default | 9400 |
| Rabbit oryCun1 | no | 50 | 3K | 3K | default | 9400 |
| Cat felCat3 | no | 50 | 3K | 3K | default | 9400 |
| Dog canFam2 | yes | 40M | 3K | 3K | default | 9400 |
| Elephant loxAfr1 | no | 50 | 3K | 3K | default | 9400 |
| Cow bosTau3 | no | 40M | 3K | 3K | default | 9400 |
| Hedgehog eriEur1 | no | 50 | 3K | 3K | default | 9400 |
| Shrew sorAra1 | no | 50 | 3K | 3K | default | 9400 |
| Tenrec echTel1 | no | 50 | 3K | 3K | default | 9400 |
| Opossum monDom4 | no | 50 | 2200 | 6000 | HoxD55 | 3400 |
| Platypus ornAna1 | no | 50 | 2200 | 6000 | HoxD55 | 3400 |
| Chicken galGal3 | yes | 40M | 2200 | 6000 | HoxD55 | 3400 |
| Lizard anoCar1 | no | 50 | 2200 | 6000 | HoxD55 | 3400 |
| X_tropicalis xenTro2 | no | 50 | 2200 | 6000 | HoxD55 | 3400 |
| Stickleback gasAcu1 | no | 50 | 2200 | 6000 | HoxD55 | 3400 |
| Zebrafish danRer5 | no | 50 | 2200 | 6000 | HoxD55 | 3400 |
| Tetraodon tetNig1 | no | 50 | 2200 | 6000 | HoxD55 | 3400 |
| Fugu fr2 | no | 50 | 2200 | 6000 | HoxD55 | 3400 |
| Medaka oryLat1 | no | 50 | 2200 | 6000 | HoxD55 | 3400 |
Additional doBlastzChainNet.pl parameters
These parameters for blastz are placed in the DEF file used by the doBlastzChainNet.pl perl script. There are some additional parameters worth discussing: 1. SEQ1* variables describe the target/reference assembly, while SEQ2* variables describe the query assembly.
If those are the same assembly, then we're doing self-alignments and will drop aligned blocks that cross the diagonal.
2. SEQn_CHUNK and SEQn_LAP determine the step size and overlap size of chunks into which large target or query sequences are to be split before alignment.
According to reasons explained in the blastz paper, SEQ1_LAP is lways set to 10000, while SEQ2_LAP is traditionaly set to 0
3. SEQn_LIMIT decides what the maximum number of sequences should be for any partitioned file (the files created in the tParts and qParts directories).
This limit only effects SEQ1 or SEQ2 when they are 2bit files. Some 2bit files have too many contigs which will result in blastz hippos (jobs taking forever compared to the other jobs). SEQ1_LIMIT defaults to 30 and SEQ2_LIMIT defaults to 100. By setting a lower limit, hippos may be avoided but more jobs will result. By setting this limit higher, fewer, longer running jobs result.
The specifics of the target and query genomes may result in very different blastz run sizes, but CHUNK and LIMIT can be used to tune the size of the blastz run.
One means of tuning is to run doBlastzChainNet.pl</COCDE> with the -stop=partition flag to test the results of the partition step until satisfied,
then continue with the -continue=blastz. To determine the number of blastz jobs that will result from a given partition, multiply the number of lines in
target.lst by the lines in the query.lst.
default blastz parameters
m=80 v=0 B=2 C=0 E=30 G=0 H=0 K=3000 L=K
M=0 O=400 P=1 R=0 T=1 W=8 X=10*(A-to-A match score)
Y=O+300*E Z=1
From the blastz usage message:
Default values are given in parentheses.
m(80M) bytes of space for trace-back information
v(0) 0: quiet; 1: verbose progress reports to stderr
B(2) 0: single strand; >0: both strands
C(0) 0: no chaining; 1: just output chain; 2: chain and extend;
3: just output HSPs
E(30) gap-extension penalty.
G(0) diagonal chaining penalty.
H(0) interpolate between alignments at threshold K = argument.
K(3000) threshold for MSPs
L(K) threshold for gapped alignments
M(0) mask any base in seq1 hit this many times; 0 = no dynamic masking
O(400) gap-open penalty.
P(1) 0: entropy not used; 1: entropy used; >1 entropy with feedback.
Q load the scoring matrix from a file.
R(0) antidiagonal chaining penalty.
T(1) 0: W-bp words; 1: 12of19; 2: 12of19 without transitions.
3: 14of22; 4: 14of22 without transitions.
W(8) word size (unused unless T=0)
X(10*(A-to-A match score)) X-drop parameter for ungapped extension.
Y(O+300E) X-drop parameter for gapped extension.
Z(1) increment between successive words in sequence 1.
The default
scoring matrix is:
The HoxD55
scoring matrix is: The mouse-rat
scoring matrix is:
A C G T A 91 -114 -31 -123 C -114 100 -125 -31 G -31 -125 100 -114 T -123 -31 -114 91
O=400 E=30
A C G T A 91 -90 -25 -100 C -90 100 -100 -25 G -25 -100 100 -90 T -100 -25 -90 91
O=400 E=30
A C G T A 56 -109 -45 -137 C -109 100 -103 -45 G -45 -103 100 -109 T -137 -45 -109 56
O=600 E=55
axtChain matrix parameters
The "medium" gap score matrix, tuned for the mouse-human distance is:
tableSize 11
smallSize 111
position 1 2 3 11 111 2111 12111 32111 72111 152111 252111
qGap 350 425 450 600 900 2900 22900 57900 117900 217900 317900
tGap 350 425 450 600 900 2900 22900 57900 117900 217900 317900
bothGap 750 825 850 1000 1300 3300 23300 58300 118300 218300 318300
The "loose" gap score matrix, tuned for the chicken-human distance is:
tablesize 11
smallSize 111
position 1 2 3 11 111 2111 12111 32111 72111 152111 252111
qGap 325 360 400 450 600 1100 3600 7600 15600 31600 56600
tGap 325 360 400 450 600 1100 3600 7600 15600 31600 56600
bothGap 625 660 700 750 900 1400 4000 8000 16000 32000 57000
the tree diagram
Created from the Hg18 28-way diagram, with the two new species, ponAbe2 and calJac1 manually inserted:
((((((((
(((Mouse_mm9:0.076274,Rat_rn4:0.084383):0.200607,
GuineaPig_cavPor2:0.202990):0.034350,
Rabbit_oryCun1:0.208548):0.014587,
((((((Human_hg18:0.005873,Chimp_panTro2:0.007668):0.013037,
Orangutan_ponAbe2:0.02):0.013037,Rhesus_rheMac2:0.031973):0.0365,
Marmoset_calJac1:0.07):0.0365,Bushbaby_otoGar1:0.151185):0.015682,
TreeShrew_tupBel1:0.162844):0.006272):0.019763,
((Shrew_sorAra1:0.248532,Hedgehog_eriEur1:0.222255):0.045693,
(((Dog_canFam2:0.101137,Cat_felCat3:0.098203):0.048213,
Horse_equCab1:0.099323):0.007287,
Cow_bosTau3:0.163945):0.012398):0.018928):0.030081,
(Armadillo_dasNov1:0.133274,(Elephant_loxAfr1:0.103030,
Tenrec_echTel1:0.232706):0.049511):0.008424):0.213469,
Opossum_monDom4:0.320721):0.088647,
Platypus_ornAna1:0.488110):0.118797,
(Chicken_galGal3:0.395136,Lizard_anoCar1:0.513962):0.093688):0.151358,
Frog_xenTro2:0.778272):0.174596,
(((Tetraodon_tetNig1:0.203933,Fugu_fr2:0.239587):0.203949,
(Stickleback_gasAcu1:0.314162,Medaka_oryLat1:0.501915):0.055354):0.346008,
Zebrafish_danRer5:0.730028):0.174596);
phastCons graph procedure
The maf files from the 30-way alignment are split for phastCons with msa_split and the following parameters:
msa_split <eachChrom>.maf -i MAF -M mm9.thisChr.fa -o SS -w 10000000,0 -I 1000 -B 5000
An attempt was made to run phyloFit on some of the smaller chromosome .ss files, but this procedure consumes too much CPU time and did not give a good result for a starting-tree to phastCons. Instead, using the starting-tree from the Hg18 28-way phastCons operation, insert the two new organisms into the tree diagram, with similar distances as seen in the failed attempt of using phyloFit on several small chromosomes, to arrive at a starting-tree.mod file:
ALPHABET: A C G T
ORDER: 0
SUBST_MOD: REV
BACKGROUND: 0.295000 0.205000 0.205000 0.295000
RATE_MAT:
-0.990634 0.178819 0.490738 0.321078
0.257325 -1.001677 0.186563 0.557790
0.706184 0.186563 -1.161873 0.269126
0.321078 0.387615 0.187019 -0.895713
TREE: (((((((((((mm9:0.076274,rn4:0.084383):0.200607,cavPor2:0.202990):0.034350,
oryCun1:0.208548):0.014587,((((((hg18:0.005873,panTro2:0.007668):0.013037,
ponAbe2:0.019969):0.013037,rheMac2:0.031973):0.013300,calJac1:0.074420):0.068818,
otoGar1:0.151185):0.015682,tupBel1:0.162844):0.006272):0.019763,
((sorAra1:0.248532,eriEur1:0.222255):0.045693,
(((canFam2:0.101137,felCat3:0.098203):0.048213,equCab1:0.099323):0.007287,
bosTau3:0.163945):0.012398):0.018928):0.030081,(dasNov1:0.133274,
(loxAfr1:0.103030,echTel1:0.232706):0.049511):0.008424):0.213469,
monDom4:0.320721):0.088647,ornAna1:0.488110):0.118797,
(galGal3:0.395136,anoCar1:0.513962):0.093688):0.151358,xenTro2:0.778272):0.174596,
(((tetNig1:0.203933,fr2:0.239587):0.203949,
(gasAcu1:0.314162,oryLat1:0.501915):0.055354):0.346008,danRer5:0.730028):0.174596);
Then, running phastCons on each .ss file with the parameters:
phastCons <oneFile>.ss starting-tree.mod --rho .31 --expected-length 45
--target-coverage .3 --quiet --seqname <chrName> --idpref <chrName>
--most-conserved <oneFile>.bed --score
Combine all the most-conserved .bed files into a single mostConserved.bed file and add lod score calculation:
cat bed/*/chr*.bed | sort -k1,1 -k2,2n | \
awk '{printf "%s\t%d\t%d\tlod=%d\t%s\n", $1, $2, $3, $5, $5;}' | \
/cluster/bin/scripts/lodToBedScore /dev/stdin > mostConserved.bed
That result is loaded for the Most Conserved track in the table phastConsElements30way. A featureBits measurement verifies a target coverage of refGene CDS near 70%:
$ featureBits mm9 -enrichment refGene:cds phastConsElements30way
refGene:cds 1.167%, phastConsElements30way 5.841%, both 0.863%, cover 73.90%, enrich 12.65x
Encoding the wiggle data from the pp files from the phastCons run:
for D in pp/chr*
do
ls $D/*.pp | sort -n -t\. -k2
done | xargs cat \
| wigEncode -noOverlap stdin phastCons30way.wig phastCons30way.wib
The sort of the files names gets them into chrom position numerical order for input to the wigEncode. Any data input to wigEncode must be in chrom position numerical order for correct encoding.
alternative phastCons graphs
Two subsets of the organisms were used to compute alternative phastCons graphs.
- Euarchontoglires - mm9,rn4,cavPor2,oryCun1,hg18,panTro2,ponAbe2,
rheMac2,calJac1,otoGar1,tupBel1
- Placentals - mm9,rn4,cavPor2,oryCun1,hg18,panTro2,ponAbe2,
rheMac2,calJac1,otoGar1,tupBel1,sorAra1,eriEur1,canFam2,felCat3,
equCab1,bosTau3,dasNov1,loxAfr1,echTel1
The same operating parameters were used during the phastCons operation.
The starting-tree.mod file for each of these was created from the overall 30-way starting-tree.mod file trimmed down with tree_doctor. Defining trees of:
Euarchontoglires:
TREE: ((((mm9:0.076274,rn4:0.084383):0.200607,cavPor2:0.202990):0.034350,
oryCun1:0.208548):0.014587,((((((hg18:0.005873,panTro2:0.007668):0.013037,
ponAbe2:0.019969):0.013037,rheMac2:0.031973):0.013300,calJac1:0.074420):0.068818,
otoGar1:0.151185):0.015682,tupBel1:0.162844):0.006272);
Placentals:
TREE: ((((((mm9:0.076274,rn4:0.084383):0.200607,cavPor2:0.202990):0.034350,
oryCun1:0.208548):0.014587,((((((hg18:0.005873,panTro2:0.007668):0.013037,
ponAbe2:0.019969):0.013037,rheMac2:0.031973):0.013300,calJac1:0.074420):0.068818,
otoGar1:0.151185):0.015682,tupBel1:0.162844):0.006272):0.019763,
((sorAra1:0.248532,eriEur1:0.222255):0.045693,
(((canFam2:0.101137,felCat3:0.098203):0.048213,equCab1:0.099323):0.007287,
bosTau3:0.163945):0.012398):0.018928):0.030081,(dasNov1:0.133274,
(loxAfr1:0.103030,echTel1:0.232706):0.049511):0.008424);
phastCons histograms
Histogram data is created from the loaded track data with the hgWiggle command:
$ hgWiggle -doHistogram -hBinSize=0.001 -hBinCount=1000 -hMinVal=0.0 -verbose=2 \
-db=mm9 phastCons30way > histogram.data 2>&1
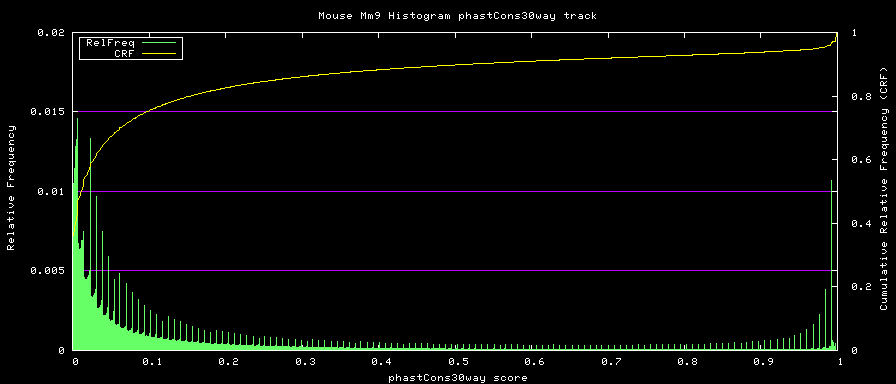 phastCons Histogram
phastCons Histogram
Euarchontoglires subset:
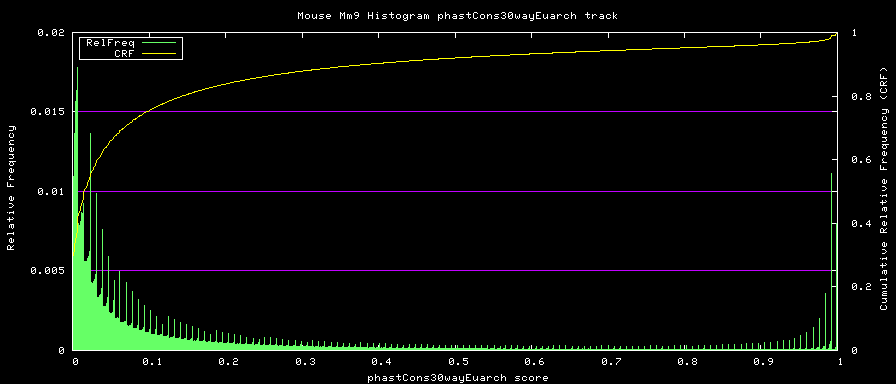 Euarchontoglires phastCons Histogram
Euarchontoglires phastCons Histogram
Placental subset:
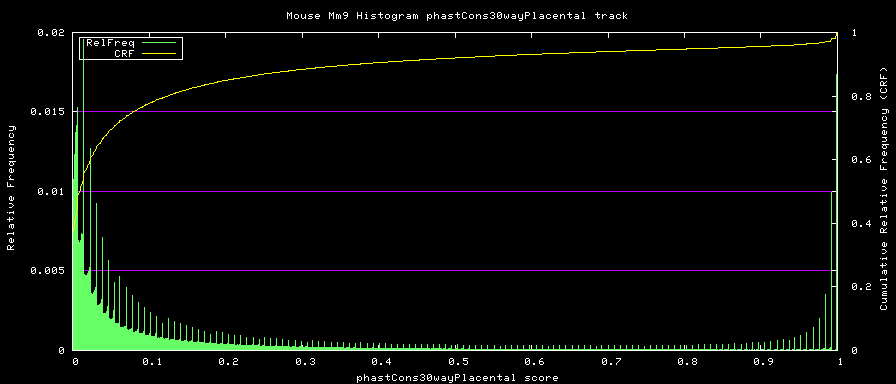 Placental phastCons Histogram
Placental phastCons Histogram