ABRF2010 Tutorial: Difference between revisions
(skeletal first draft.) |
(Links changed from .soe or .cse to .gi) |
||
| (41 intermediate revisions by 6 users not shown) | |||
| Line 1: | Line 1: | ||
Presented by Angie Hinrichs and Robert Kuhn March 20, 2010 as part of the workshop <em>Tools to Facilitate Management, Analysis and Visualization of 2nd Generation Sequencing Data</em> at [http://www.abrf.org/ ABRF 2010]. | |||
Presented March 20, 2010 as part of the workshop | |||
== Intro: Basic Genome Browser navigation== | |||
== Genome Graphs with .sgr data == | Here we will explore the Genome Browser to get familiar with moving around the landscape and load a simple Custom Track | ||
===Navigation -- find a gene=== | |||
There are several ways to get started. We will start with a clean "cart", select a genome assembly and enter a gene name. | |||
====Steps==== | |||
# Go to http://genome.ucsc.edu and hit the button "Genome Browser at the upper left in the blue bar. | |||
# Click here to reset (below the pulldown menus). This puts us all at the same place. | |||
# Select assembly hg18 (has richer annotations than the newer hg19, which is still being populated with data sets). | |||
# In the "gene" box, slowly type "PIGP." You will see the choices of gene names appear in the pulldown as you type. | |||
# Got to the gene, PIGP, by hitting the "jump" button. | |||
===Navigation -- zooming, panning and controlling data tracks=== | |||
To get around in the browser view, you can move to any continuously variable region of your choosing. | |||
====Steps==== | |||
# Above the browser image, hit the "zoom out 3x" button to see the region around the gene. | |||
# Data in the various "tracks" are drawn from separate tables in the browser database. | |||
# Note the spikes of conserved sequence among the species in the Conservation track. | |||
# Position your cursor at the top of the image in the scale bar above one of the exons. Hold down the mouse button and drag the mouse right or left to highlight the exon. Release the button to expand the exon to full screen. | |||
# We'll turn off some of the default tracks to simplify the view. As we are concerned about next-gen sequencing in this workshop, we'll turn off the Array track. Near the bottom of the browser image, find the track, "SNP Genotyping Arrays" and click the little minibutton to the left side of the track. | |||
# On the configuration page, we could turn on/off any of the subtracks. Select "hide" from the pulldown and Submit. Each data track has configuration options accessible via the little button. | |||
# Below the image, in the track controls, find these tracks and select "hide": RefGene, Human mRNAs, Spliced ESTs, Repeat Masker. Select "Refresh" from any blue bar. | |||
# The UCSC Genes Track, SNPs, and Conservation are still displayed. Note the little chevron arrows on UCSC Gene, PIGP, showing that it is transcribed from the negative strand. Click it to see rich information about the gene. | |||
# Return to the browser via the link the blue bar at the top. | |||
===Custom Tracks -- add your own data=== | |||
We will load a simple Custom Track to see how user-supplied data can be added to the data already in the browser. | |||
====Steps==== | |||
# Below the browser image, select "add custom tracks." | |||
# In the upper box, type (or paste): | |||
track name="test custom track" visibility=pack | |||
chr21 10000000 10050000 | |||
chr21 10025000 10100000 | |||
This is BED 3 format: chrom, chromStart, chromEnd | |||
# Select "Submit." | |||
# At the browser, select "zoom out 10x" | |||
== Tutorial: Viewing sequencing data in the UCSC Genome Browser == | |||
This page serves as a step-by-step guide to the demo of UCSC Genome Browser tools presented at the workshop. Earlier in the workshop, Charlie Nicolet shared some sequencing and analysis results on chromosome 21 for ChIP-Seq of RNA polymerase II to HeLa cells. Thanks to Charlie for allowing us to post copies of his data and use them as a basis for this tutorial. | |||
In case internet fails at the workshop, slides will be used ([[Image:ABRF2010 UCSC NGS.ppt]] | [[Image:ABRF2010 UCSC NGS.pdf]]). | |||
=== Genome Graphs with .sgr data === | |||
Some of the result files from the ChIPSeq toolset that Charlie Nicolet showed us earlier are in the .sgr format, i.e. tab-separated text that specifies chromosome, position and a numeric signal value. This format can be uploaded to UCSC's Genome Graphs tool for genome-at-a-glance view of the data. | Some of the result files from the ChIPSeq toolset that Charlie Nicolet showed us earlier are in the .sgr format, i.e. tab-separated text that specifies chromosome, position and a numeric signal value. This format can be uploaded to UCSC's Genome Graphs tool for genome-at-a-glance view of the data. | ||
# genome.ucsc.edu | ====Steps==== | ||
# 7th link | |||
# | # In a new web browser window or tab, go to '''<nowiki>http://genome.ucsc.edu/</nowiki>''' . | ||
# upload | # On the left side of the page, there is a blue sidebar. Click on "Genome Graphs", the 7th link from the top of the sidebar. | ||
# should see confirmation screen | # In the assembly menu, change the selection to "Mar. 2006 (NCBI36/hg18)". | ||
# Genome Graphs doesn't show the data until you select it. | # Click the upload button. | ||
# In the input labeled "name of data set", type or paste "redbin". | |||
# Paste this URL: '''<nowiki>http://genome-test.gi.ucsc.edu/ABRF2010/chr21_extended.txt_redbin.sgr</nowiki>''' in the box labeled "Paste URLs or data", and click submit. | |||
# Note the track at the top labeled "redbin 1" -- that is the data shown in Genome Graphs. | # You should see confirmation screen; click OK to return to the Genome Graphs display. | ||
# Now you should see a layout of human chromosomes. Genome Graphs doesn't show the data until you select it. In the graph menu on the left, change "--nothing--" to "redbin 1". Now the data appear -- in this case, for chr21 only. | |||
[[Image:ABRF2010_ggData.png|border|link=http://genome.ucsc.edu/cgi-bin/hgGenome?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_GG|catch up]] | |||
If you don't see blue data appearing over chromosome 21, go to http://genome.ucsc.edu/cgi-bin/hgGenome?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_GG to catch up. | |||
Now we will use Genome Graphs to navigate to the Genome Browser. | |||
# Click on a peak to jump to the Genome Browser, displaying a 1,000,000-base region of the genome corresponding to the location you clicked in Genome Graphs. | |||
# Note the track at the top labeled "redbin 1" on the left and "User Supplied Track 1" above -- that is the data shown in Genome Graphs. Click on the top label "User Supplied Track 1" to make the peaks taller and more visible. In general, peaks correspond to upstream regions of genes, which makes sense for RNA polymerase II ChIP-seq. | |||
[[Image:ABRF2010 redbin 1.png|border|link=http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_redbin_1|catch up]] | |||
If you don't see the above, go to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_redbin_1 to catch up. | |||
== Transforming .sgr into bedGraph for Genome Browser viewing == | === Transforming .sgr into bedGraph for Genome Browser viewing === | ||
The Genome Browser can draw a somewhat nicer display of .sgr data, if we do a simple transform of the .sgr data into a similar format called bedGraph. bedGraph has both start and end genomic coordinates, so scores can be drawn over regions as opposed to single bases. Here is a command that translates .sgr to bedGraph, expanding each data point to cover a 30-base region: | The Genome Browser can draw a somewhat nicer display of .sgr data, if we do a simple transform of the .sgr data into a similar format called bedGraph. bedGraph has both start and end genomic coordinates, so scores can be drawn over regions as opposed to single bases. Here is a command that translates .sgr to bedGraph, expanding each data point to cover a 30-base region: | ||
perl -wpe '@w=split("\t"); $_ = "$w[0]\t" . ($w[1]-1) . "\t" . ($w[1]+29) . "\t$w[2]";' \ | perl -wpe '@w=split("\t"); $_ = "$w[0]\t" . ($w[1]-1) . "\t" . ($w[1]+29) . "\t$w[2]";' \ | ||
chr21_extended.txt_redbin.sgr > | chr21_extended.txt_redbin.sgr > redbin.bed | ||
If you would like to show only datapoints above a certain threshold, say 5, you can make the command slightly more complicated: | If you would like to show only datapoints above a certain threshold, say 5, you can make the command slightly more complicated: | ||
perl -wpe '@w=split("\t"); $_ = "$w[0]\t" . ($w[1]-1) . "\t" . ($w[1]+29) . "\t$w[2]"; \ | perl -wpe '@w=split("\t"); $_ = "$w[0]\t" . ($w[1]-1) . "\t" . ($w[1]+29) . "\t$w[2]"; \ | ||
if ($w[2] < | if ($w[2] < 15) {$_ = "";}' \ | ||
chr21_extended.txt_redbin.sgr > | chr21_extended.txt_redbin.sgr > redbinGt15.bed | ||
This line can be added to the beginning of redbinGt15.bed, in order to add labels to the track display and configure default appearance (see http://genome.ucsc.edu/goldenPath/help/bedgraph.html for more information about settings). A line break is used for readability here, but must be removed if you are going to edit this track line into a file: | |||
track name=redbinGt15 description="redbin items with score >= 15, expanded to 30 bases" \ | |||
type=bedGraph viewLimits=0:100 autoScale=off maxHeightPixels=128:20:11 | |||
The file has been saved here: http://genome-test.gi.ucsc.edu/ABRF2010/redbinGt15.bed . | |||
====Steps==== | |||
# In the Genome Browser, click the "manage custom tracks" button below the image. | # In the Genome Browser, click the "manage custom tracks" button below the image. | ||
# | # Click on "add custom tracks" | ||
# You should see a confirmation page like this ('' | # Paste these two lines into the box labeled "Paste URLs or data" and click submit: | ||
# | '''<nowiki>http://genome-test.gi.ucsc.edu/ABRF2010/redbinGt15.bed</nowiki>''' | ||
# Now the new custom track shows grayscale representations of the score. You can click on the track title ("redbin items with score > | '''browser position chr21:42,886,797-43,886,796''' | ||
# You should see a confirmation page like this (''image'') | |||
# To go to the genome browser, you can click on "Genome Browser" in the top blue bar, or click on "chr21" in the Pos column of the table showing your custom tracks, or on the "go to genome browser" button. Note: in the absence of a "browser position" line, only the top blue bar link will return you to the position you were looking at before. | |||
# Now the new custom track shows grayscale representations of the score. You can click on the track title ("redbin items with score > 15") to expand the display to bar graph, which should look a lot like the "redbin 1" track created in Genome Graphs. | |||
[[Image:ABRF2010_redbinGt15.png|border|link=http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_redbinGt15|catch up]] | |||
If you don't see two tracks labeled "redbin 1" and "redbinGt15" over the UCSC Genes track, go to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_redbinGt15 to catch up. | |||
=== Comparing with UCSC-hosted data === | |||
Now we will take a look at some ChIP-seq data from the ENCODE project. The "Yale TFBS" track in the Regulation section combines ChIP-seq data from several labs, and it happens to include data for RNA polymerase II ("Pol2") in HeLa-S3 cells, like Charlie Nicolet's chr21 data. We can visually compare the two datasets to see how well they correspond. | |||
== | ====Steps==== | ||
# In the Genome Browser, scroll down below the image to the | # In the Genome Browser, scroll down below the image to the Regulation section. Click the "+" button on the left side of the blue "Regulation" header to expand. | ||
# Click on the link titled "Yale TFBS". This will take you to the track controls. | # Click on the link titled "Yale TFBS". This will take you to the track controls. | ||
# In the large table under "Select subtracks by cell line and factor", | # In the large table under "Select subtracks by cell line and factor", | ||
# Hit the "-" button in upper left corner to deselect all subtracks. Note that HeLa-S3 cell line is in 3rd column of checkboxes. | # Hit the "-" button in upper left corner to deselect all subtracks. Note that HeLa-S3 cell line is in 3rd column of checkboxes. | ||
# Scroll down to factor Pol2. select the 3rd check box. Only HeLa/Pol2 is selected for display. | # Scroll down to factor Pol2. select the 3rd check box. Only HeLa/Pol2 is selected for display. | ||
# Scroll back up to the top of the page. Change visibility to " | # Scroll back up to the top of the page. Change visibility to "full" and click submit. | ||
# In the Genome Browser, HeLa/Pol2 peaks and signal appear below "Spliced ESTs". '' | # In the Genome Browser, HeLa/Pol2 peaks and signal appear below "Spliced ESTs". | ||
The intervening tracks make it a little difficult to compare the ENCODE data with Charlie's, so we will adjust the display to see only the tracks we want to compare. | |||
# Click the "hide all" button under the image. | |||
# In the Custom Tracks section below the image, change the visibilities of redbin 1 and redbinGt15 to "full". | |||
# In the Genes and Gene Prediciton Tracks section, change the visibility of UCSC Genes to "dense". | |||
# In the Regulation section, change the visibility of Yale TFBS to "full". | |||
# Click the "refresh" button on any blue section header. | |||
[[Image:ABRF2010_TFBS.png|border|link=http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_TFBS|catch up]] | |||
If you don't see a clear correspondence between the track on the bottom and the tracks on the top, go to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_TFBS to catch up. | |||
=== Viewing alignments in BAM format === | |||
Charlie Nicolet has shared the ELAND export file that accompanies chr21_extended.txt, [http://genome-test.gi.ucsc.edu/ABRF2010/chr21_export.txt chr21_export.txt]. This can be transformed into [http://samtools.sourceforge.net/ BAM] for viewing in the UCSC Genome Browser. The [http://samtools.sourceforge.net/ samtools] package includes a script that converts ELAND export files into SAM (the text version of BAM). Export files often have fasta names instead of sequence names, e.g. "chr21.fa" instead of "chr21", so the ".fa" must be removed: | |||
export2sam.pl --read1=chr21_export.txt \ | |||
| perl -wpe 's/(chr.*)\.fa/$1/' \ | |||
> chr21.sam | |||
Edit chr21.sam to add these header lines at the beginning (note: the words must be separated by tab characters, not spaces): | |||
@HD VN:1.0 GO:none SO:coordinate | |||
@SQ SN:chr21 LN:46944323 | |||
Note that current versions of samtools do not require the @SQ fields, you can also add the -t option to the "samtools view" command below and specify a chromSizes.txt file in that command (from the chromInfo table, e.g. using the fetchChromSizes script). | |||
Use samtools to convert SAM to BAM, sort the BAM, and index the sorted BAM (see http://genome.ucsc.edu/goldenPath/help/bam.html for more info): | |||
samtools view -bSo chr21.unsorted.bam chr21.sam | |||
samtools sort chr21.unsorted.bam chr21 | |||
samtools index chr21.bam | |||
Now the sorted chr21.bam and its index file chr21.bam.bai must be put on a web server (ftp, http, or https). For example, if you create files my.bam and my.bam.bai, and you have a personal web directory <nowiki>http://my.edu/mylab/~me/</nowiki>, you can copy or link the files to <nowiki>http://my.edu/mylab/~me/my.bam</nowiki> and <nowiki>http://mylab/~me/my.bam.bai</nowiki>. Then you would use only the first URL (<nowiki>http://my.edu/mylab/~me/my.bam</nowiki>) in a track definition line like the one below. For this example, we will use <nowiki>http://genome-test.gi.ucsc.edu/ABRF2010/chr21.bam</nowiki> . | |||
====Steps==== | |||
# In the Genome Browser, click the "manage custom tracks" buttom below the image. | |||
# Click the "add custom tracks" button. | |||
# Paste this text into the box: | |||
'''track name=chr21_export type=bam bigDataUrl=<nowiki>http://genome-test.gi.ucsc.edu/ABRF2010/chr21.bam</nowiki> visibility=pack''' | |||
# Click submit. Click "Genome Browser" in the blue top bar. Now you should see an almost-solid dark blue bar with a few dark red stripes, labeled "chr21_export" above and to the left. | |||
[[Image:ABRF2010_chr21_export_1MB.png|border|link=http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_chr21_export_1MB|catch up]] | |||
If you don't see the above, got to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_chr21_export_1MB to catch up. There are so many alignments in this 1MB window that the items can't be drawn individually -- the image would become too tall. Let's view a smaller genomic region to get a better look at the alignments. | |||
# Click and drag on the base position track to select a window around the couple largest peaks. (''If it doesn't work for you, paste '''chr21:43,169,278-43,191,176''' into the position box and click "jump".'') | |||
# Now we are viewing about 22,000 bases covering two peaks, and there are still so many alignments that the track is forced into "squish" display mode: items are shown at half-height and read names are suppressed. Let's zoom in again, to the right side of the left peak ''(or jump to position '''chr21:43,172,778-43,173,354'''). | |||
# Even when viewing 577 bases, there is still quite a pileup of read alignments to view. The read names take up a lot of room, so let's hide them. Click on the light blue bar to the left of the chr21_export track, to go to track controls. | |||
# There is a checkbox to the right of "Display read names" -- click to clear the checkbox, and then click submit. | |||
# Mismatches appear as light red tickmarks. They are a bit easier to see if the items are drawn in grayscale, so again click on the light blue bar on the left to return to track controls. | |||
# Under "additional coloring modes", select "Use gray for [alignment quality]", and click submit. | |||
# Note the trend for mismatches to appear more often in alignments with lower quality, and also the relative frequency of lower-quality reads at the edge of the peak. | |||
[[Image:ABRF2010 chr21 export grayQual.png|border|link=|catch up]] | |||
If you don't see the above, go to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_chr21_export_grayQual to catch up. | |||
# Zoom in once again to focus on the region with the most mismatching items ('''chr21:43,172,834-43,172,931'''). Now we see the exact mismatches between reference and read bases. | |||
# Let's take a look at base qualities. Once again, click the light blue bar on the left to return to track controls. | |||
# Change the selection to "Use gray for [base qualities]", and click submit. Some of the mismatches, but not all, almost disappear due to the light shades used for low quality bases. | |||
[[Image:ABRF2010 chr21 export baseQual.png|border|link=http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_chr21_export_baseQual|catch up]] | |||
If you don't see the above, go to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_chr21_export_baseQual to catch up. | |||
====Extra: Finding variants in alignment display==== | |||
In some regions, stacks of red tickmarks (mismatches) indicate HeLa variants with respect to the reference genome assembly. Here are several examples. | |||
# Paste '''chr21:37,366,724-37,366,992''' into the position box and click "jump". | |||
# To compare with variants in dbSNP, scroll down to the Variation section and change the visibility of SNPs (130) to pack. Click "refresh" on any blue section header. | |||
# The SNP track appears at the bottom of the image. SNPs classified "coding non-synonymous" by dbSNP are colored red. ''(If you don't see that, click '''<nowiki>http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_chr21_export_SNPs</nowiki>''' to catch up.)'' | |||
# To see a variant not in dbSNP, paste '''chr21:37,367,831-37,367,845'''. | |||
# To see a heterozygous variant adjacent to a dbSNP 3-base indel, paste '''chr21:37,367,478-37,367,492'''. | |||
# This 110-base region contains three know variations from the reference assembly: '''chr21:37,366,860-37,366,969'''. | |||
== | == Where to learn more == | ||
* [http://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html Help] link in blue top bar of any page | |||
* [http://genome.ucsc.edu/FAQ/ FAQ] (Frequently asked questions) | |||
* mailto:genome@soe.ucsc.edu and list archives | |||
* [[Main_Page|http://genomewiki.ucsc.edu/]] (this site) | |||
* [http://www.openhelix.com/downloads/ucsc/ucsc_home.shtml OpenHelix] offers free tutorials and reference card PDFs. | |||
[[Category:Browser Linked]] | |||
Latest revision as of 20:34, 24 September 2018
Presented by Angie Hinrichs and Robert Kuhn March 20, 2010 as part of the workshop Tools to Facilitate Management, Analysis and Visualization of 2nd Generation Sequencing Data at ABRF 2010.
Here we will explore the Genome Browser to get familiar with moving around the landscape and load a simple Custom Track
There are several ways to get started. We will start with a clean "cart", select a genome assembly and enter a gene name.
Steps
- Go to http://genome.ucsc.edu and hit the button "Genome Browser at the upper left in the blue bar.
- Click here to reset (below the pulldown menus). This puts us all at the same place.
- Select assembly hg18 (has richer annotations than the newer hg19, which is still being populated with data sets).
- In the "gene" box, slowly type "PIGP." You will see the choices of gene names appear in the pulldown as you type.
- Got to the gene, PIGP, by hitting the "jump" button.
To get around in the browser view, you can move to any continuously variable region of your choosing.
Steps
- Above the browser image, hit the "zoom out 3x" button to see the region around the gene.
- Data in the various "tracks" are drawn from separate tables in the browser database.
- Note the spikes of conserved sequence among the species in the Conservation track.
- Position your cursor at the top of the image in the scale bar above one of the exons. Hold down the mouse button and drag the mouse right or left to highlight the exon. Release the button to expand the exon to full screen.
- We'll turn off some of the default tracks to simplify the view. As we are concerned about next-gen sequencing in this workshop, we'll turn off the Array track. Near the bottom of the browser image, find the track, "SNP Genotyping Arrays" and click the little minibutton to the left side of the track.
- On the configuration page, we could turn on/off any of the subtracks. Select "hide" from the pulldown and Submit. Each data track has configuration options accessible via the little button.
- Below the image, in the track controls, find these tracks and select "hide": RefGene, Human mRNAs, Spliced ESTs, Repeat Masker. Select "Refresh" from any blue bar.
- The UCSC Genes Track, SNPs, and Conservation are still displayed. Note the little chevron arrows on UCSC Gene, PIGP, showing that it is transcribed from the negative strand. Click it to see rich information about the gene.
- Return to the browser via the link the blue bar at the top.
Custom Tracks -- add your own data
We will load a simple Custom Track to see how user-supplied data can be added to the data already in the browser.
Steps
- Below the browser image, select "add custom tracks."
- In the upper box, type (or paste):
track name="test custom track" visibility=pack chr21 10000000 10050000 chr21 10025000 10100000
This is BED 3 format: chrom, chromStart, chromEnd
- Select "Submit."
- At the browser, select "zoom out 10x"
Tutorial: Viewing sequencing data in the UCSC Genome Browser
This page serves as a step-by-step guide to the demo of UCSC Genome Browser tools presented at the workshop. Earlier in the workshop, Charlie Nicolet shared some sequencing and analysis results on chromosome 21 for ChIP-Seq of RNA polymerase II to HeLa cells. Thanks to Charlie for allowing us to post copies of his data and use them as a basis for this tutorial.
In case internet fails at the workshop, slides will be used (File:ABRF2010 UCSC NGS.ppt | File:ABRF2010 UCSC NGS.pdf).
Genome Graphs with .sgr data
Some of the result files from the ChIPSeq toolset that Charlie Nicolet showed us earlier are in the .sgr format, i.e. tab-separated text that specifies chromosome, position and a numeric signal value. This format can be uploaded to UCSC's Genome Graphs tool for genome-at-a-glance view of the data.
Steps
- In a new web browser window or tab, go to http://genome.ucsc.edu/ .
- On the left side of the page, there is a blue sidebar. Click on "Genome Graphs", the 7th link from the top of the sidebar.
- In the assembly menu, change the selection to "Mar. 2006 (NCBI36/hg18)".
- Click the upload button.
- In the input labeled "name of data set", type or paste "redbin".
- Paste this URL: http://genome-test.gi.ucsc.edu/ABRF2010/chr21_extended.txt_redbin.sgr in the box labeled "Paste URLs or data", and click submit.
- You should see confirmation screen; click OK to return to the Genome Graphs display.
- Now you should see a layout of human chromosomes. Genome Graphs doesn't show the data until you select it. In the graph menu on the left, change "--nothing--" to "redbin 1". Now the data appear -- in this case, for chr21 only.
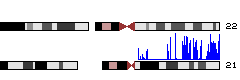
If you don't see blue data appearing over chromosome 21, go to http://genome.ucsc.edu/cgi-bin/hgGenome?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_GG to catch up.
Now we will use Genome Graphs to navigate to the Genome Browser.
- Click on a peak to jump to the Genome Browser, displaying a 1,000,000-base region of the genome corresponding to the location you clicked in Genome Graphs.
- Note the track at the top labeled "redbin 1" on the left and "User Supplied Track 1" above -- that is the data shown in Genome Graphs. Click on the top label "User Supplied Track 1" to make the peaks taller and more visible. In general, peaks correspond to upstream regions of genes, which makes sense for RNA polymerase II ChIP-seq.
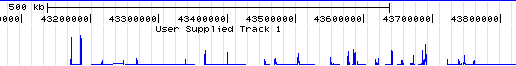
If you don't see the above, go to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_redbin_1 to catch up.
Transforming .sgr into bedGraph for Genome Browser viewing
The Genome Browser can draw a somewhat nicer display of .sgr data, if we do a simple transform of the .sgr data into a similar format called bedGraph. bedGraph has both start and end genomic coordinates, so scores can be drawn over regions as opposed to single bases. Here is a command that translates .sgr to bedGraph, expanding each data point to cover a 30-base region:
perl -wpe '@w=split("\t"); $_ = "$w[0]\t" . ($w[1]-1) . "\t" . ($w[1]+29) . "\t$w[2]";' \
chr21_extended.txt_redbin.sgr > redbin.bed
If you would like to show only datapoints above a certain threshold, say 5, you can make the command slightly more complicated:
perl -wpe '@w=split("\t"); $_ = "$w[0]\t" . ($w[1]-1) . "\t" . ($w[1]+29) . "\t$w[2]"; \
if ($w[2] < 15) {$_ = "";}' \
chr21_extended.txt_redbin.sgr > redbinGt15.bed
This line can be added to the beginning of redbinGt15.bed, in order to add labels to the track display and configure default appearance (see http://genome.ucsc.edu/goldenPath/help/bedgraph.html for more information about settings). A line break is used for readability here, but must be removed if you are going to edit this track line into a file:
track name=redbinGt15 description="redbin items with score >= 15, expanded to 30 bases" \
type=bedGraph viewLimits=0:100 autoScale=off maxHeightPixels=128:20:11
The file has been saved here: http://genome-test.gi.ucsc.edu/ABRF2010/redbinGt15.bed .
Steps
- In the Genome Browser, click the "manage custom tracks" button below the image.
- Click on "add custom tracks"
- Paste these two lines into the box labeled "Paste URLs or data" and click submit:
http://genome-test.gi.ucsc.edu/ABRF2010/redbinGt15.bed browser position chr21:42,886,797-43,886,796
- You should see a confirmation page like this (image)
- To go to the genome browser, you can click on "Genome Browser" in the top blue bar, or click on "chr21" in the Pos column of the table showing your custom tracks, or on the "go to genome browser" button. Note: in the absence of a "browser position" line, only the top blue bar link will return you to the position you were looking at before.
- Now the new custom track shows grayscale representations of the score. You can click on the track title ("redbin items with score > 15") to expand the display to bar graph, which should look a lot like the "redbin 1" track created in Genome Graphs.
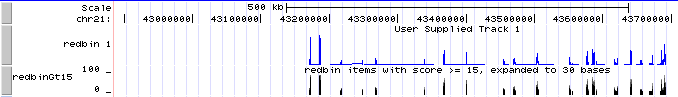
If you don't see two tracks labeled "redbin 1" and "redbinGt15" over the UCSC Genes track, go to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_redbinGt15 to catch up.
Comparing with UCSC-hosted data
Now we will take a look at some ChIP-seq data from the ENCODE project. The "Yale TFBS" track in the Regulation section combines ChIP-seq data from several labs, and it happens to include data for RNA polymerase II ("Pol2") in HeLa-S3 cells, like Charlie Nicolet's chr21 data. We can visually compare the two datasets to see how well they correspond.
Steps
- In the Genome Browser, scroll down below the image to the Regulation section. Click the "+" button on the left side of the blue "Regulation" header to expand.
- Click on the link titled "Yale TFBS". This will take you to the track controls.
- In the large table under "Select subtracks by cell line and factor",
- Hit the "-" button in upper left corner to deselect all subtracks. Note that HeLa-S3 cell line is in 3rd column of checkboxes.
- Scroll down to factor Pol2. select the 3rd check box. Only HeLa/Pol2 is selected for display.
- Scroll back up to the top of the page. Change visibility to "full" and click submit.
- In the Genome Browser, HeLa/Pol2 peaks and signal appear below "Spliced ESTs".
The intervening tracks make it a little difficult to compare the ENCODE data with Charlie's, so we will adjust the display to see only the tracks we want to compare.
- Click the "hide all" button under the image.
- In the Custom Tracks section below the image, change the visibilities of redbin 1 and redbinGt15 to "full".
- In the Genes and Gene Prediciton Tracks section, change the visibility of UCSC Genes to "dense".
- In the Regulation section, change the visibility of Yale TFBS to "full".
- Click the "refresh" button on any blue section header.
![]()
If you don't see a clear correspondence between the track on the bottom and the tracks on the top, go to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_TFBS to catch up.
Viewing alignments in BAM format
Charlie Nicolet has shared the ELAND export file that accompanies chr21_extended.txt, chr21_export.txt. This can be transformed into BAM for viewing in the UCSC Genome Browser. The samtools package includes a script that converts ELAND export files into SAM (the text version of BAM). Export files often have fasta names instead of sequence names, e.g. "chr21.fa" instead of "chr21", so the ".fa" must be removed:
export2sam.pl --read1=chr21_export.txt \
| perl -wpe 's/(chr.*)\.fa/$1/' \
> chr21.sam
Edit chr21.sam to add these header lines at the beginning (note: the words must be separated by tab characters, not spaces):
@HD VN:1.0 GO:none SO:coordinate @SQ SN:chr21 LN:46944323
Note that current versions of samtools do not require the @SQ fields, you can also add the -t option to the "samtools view" command below and specify a chromSizes.txt file in that command (from the chromInfo table, e.g. using the fetchChromSizes script).
Use samtools to convert SAM to BAM, sort the BAM, and index the sorted BAM (see http://genome.ucsc.edu/goldenPath/help/bam.html for more info):
samtools view -bSo chr21.unsorted.bam chr21.sam samtools sort chr21.unsorted.bam chr21 samtools index chr21.bam
Now the sorted chr21.bam and its index file chr21.bam.bai must be put on a web server (ftp, http, or https). For example, if you create files my.bam and my.bam.bai, and you have a personal web directory http://my.edu/mylab/~me/, you can copy or link the files to http://my.edu/mylab/~me/my.bam and http://mylab/~me/my.bam.bai. Then you would use only the first URL (http://my.edu/mylab/~me/my.bam) in a track definition line like the one below. For this example, we will use http://genome-test.gi.ucsc.edu/ABRF2010/chr21.bam .
Steps
- In the Genome Browser, click the "manage custom tracks" buttom below the image.
- Click the "add custom tracks" button.
- Paste this text into the box:
track name=chr21_export type=bam bigDataUrl=http://genome-test.gi.ucsc.edu/ABRF2010/chr21.bam visibility=pack
- Click submit. Click "Genome Browser" in the blue top bar. Now you should see an almost-solid dark blue bar with a few dark red stripes, labeled "chr21_export" above and to the left.
![]()
If you don't see the above, got to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_chr21_export_1MB to catch up. There are so many alignments in this 1MB window that the items can't be drawn individually -- the image would become too tall. Let's view a smaller genomic region to get a better look at the alignments.
- Click and drag on the base position track to select a window around the couple largest peaks. (If it doesn't work for you, paste chr21:43,169,278-43,191,176 into the position box and click "jump".)
- Now we are viewing about 22,000 bases covering two peaks, and there are still so many alignments that the track is forced into "squish" display mode: items are shown at half-height and read names are suppressed. Let's zoom in again, to the right side of the left peak (or jump to position chr21:43,172,778-43,173,354).
- Even when viewing 577 bases, there is still quite a pileup of read alignments to view. The read names take up a lot of room, so let's hide them. Click on the light blue bar to the left of the chr21_export track, to go to track controls.
- There is a checkbox to the right of "Display read names" -- click to clear the checkbox, and then click submit.
- Mismatches appear as light red tickmarks. They are a bit easier to see if the items are drawn in grayscale, so again click on the light blue bar on the left to return to track controls.
- Under "additional coloring modes", select "Use gray for [alignment quality]", and click submit.
- Note the trend for mismatches to appear more often in alignments with lower quality, and also the relative frequency of lower-quality reads at the edge of the peak.

If you don't see the above, go to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_chr21_export_grayQual to catch up.
- Zoom in once again to focus on the region with the most mismatching items (chr21:43,172,834-43,172,931). Now we see the exact mismatches between reference and read bases.
- Let's take a look at base qualities. Once again, click the light blue bar on the left to return to track controls.
- Change the selection to "Use gray for [base qualities]", and click submit. Some of the mismatches, but not all, almost disappear due to the light shades used for low quality bases.
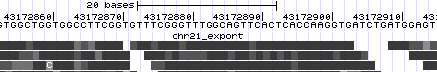
If you don't see the above, go to http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_chr21_export_baseQual to catch up.
Extra: Finding variants in alignment display
In some regions, stacks of red tickmarks (mismatches) indicate HeLa variants with respect to the reference genome assembly. Here are several examples.
- Paste chr21:37,366,724-37,366,992 into the position box and click "jump".
- To compare with variants in dbSNP, scroll down to the Variation section and change the visibility of SNPs (130) to pack. Click "refresh" on any blue section header.
- The SNP track appears at the bottom of the image. SNPs classified "coding non-synonymous" by dbSNP are colored red. (If you don't see that, click http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=AngieHinrichs&hgS_otherUserSessionName=ABRF2010_chr21_export_SNPs to catch up.)
- To see a variant not in dbSNP, paste chr21:37,367,831-37,367,845.
- To see a heterozygous variant adjacent to a dbSNP 3-base indel, paste chr21:37,367,478-37,367,492.
- This 110-base region contains three know variations from the reference assembly: chr21:37,366,860-37,366,969.
Where to learn more
- Help link in blue top bar of any page
- FAQ (Frequently asked questions)
- mailto:genome@soe.ucsc.edu and list archives
- http://genomewiki.ucsc.edu/ (this site)
- OpenHelix offers free tutorials and reference card PDFs.